Kaj je Marfanov sindrom?
Marfanov sindrom opisuje kompleksno dedno motnjo vezivnega tkiva, ki večinoma prizadene oči, kardiovaskularni sistem in skeletni mišični sistem. Glede na to, da je vsak organ sestavljen iz veznega tkiva, lahko Marfanov sindrom uniči in močno ovira rast in delovanje vsakega anatomskega mesta.
Sindrom se prenaša kot avtosomno dominantna lastnost: zato se soočamo z resno genetsko boleznijo, ki ima izredno spremenljivo fenotipsko izražanje (napake se lahko zelo razlikujejo od družine do družine ali od bolnika do bolnika).
Kar povzroča Marfanov sindrom je sprememba gena FBN1 (na kromosomu 15), ki kodira fibrilin-1, zelo pomemben vezni glikoprotein, ki predstavlja strukturno podporo mikrofibrilom.
Mikrofibrile: sestavljene iz fibrilina, mikrofibrile so prisotne v zunajceličnem matriksu, v katerem tvorijo mrežo za odlaganje elastina v elastičnih vlaknih. Čeprav je v telesu prisotna povsod, mikrofibrile v glavnem prežijo v aorti, ligamentih in zonilih cilijarnih teles (na ravni očesa).
Kot avtosomno dominantna bolezen Marfanov sindrom prizadene le otroke, ki so podedovali gen FBN-1, ki ga spreminjajo oba starša. Kljub temu je v enem primeru od štirih bolezen posledica spontanih mutacij pri bolnikih, ki nimajo družinske anamneze.
Ime bolezni prihaja od francoskega pediatra, ki ga je prvič opisal leta 1896 (A. Marfan), po katerem je moral do leta 1991 počakati na identifikacijo spremenjenega gena, ki je bil vključen v simptomatsko manifestacijo: odkritelj je bil F. Ramirez.
Oglejte si video
X Oglejte si videoposnetek na youtubevzroki
Omenili smo, da je Marfanov sindrom takojšen izraz mutacije gena, ki kodira fibrilin-1. Poskusimo poglobiti temo, da bi razumeli, kateri mehanizem je vzpostavljen za sprožitev sindroma.
FIBRILLINA 1 je glikoproteinski del elastina, ki je bistven za zagotavljanje in vzdrževanje elastičnosti in moči tkiva. V fizioloških pogojih se fibrilin 1 veže na drug protein, znan kot TGF-beta (ali transformirajoči rastni beta faktor). Zdi se, da je TGF-beta vključen v škodljive procese, ki vplivajo na gladke mišice žil in na zunajcelični matriks. Izhajajoč iz teh predpostavk so nekateri avtorji prepričani, da je Marfanov sindrom poleg mutacije gena FBN-1 posledica tudi prevelike količine TGF-beta, zlasti v aorti, v srčnih valjih in v pljučih.
incidenca
Ocenjuje se, da Marfanov sindrom prizadene 1 osebo na vsakih 3.000-5.000 rojstev in se ne kaže jasno med moškimi in ženskami, brez nagnjenja k natančni pasmi. Statistični podatki kažejo, da ima 75% bolnikov pozitivno družinsko zgodovino; v preostalih 25% je vzrok le sporadične mutacije, ki so na nek način povezane z visoko starostjo očeta v času zanositve.
Otroci z zelo hudimi oblikami Marfanovega sindroma imajo pričakovano življenjsko dobo manj kot eno leto.
Pred razvojem kirurških strategij odprtega srca je imela večina bolnikov z Marfanovim sindromom povprečno življenjsko dobo 32 let; zaradi nenehnega izboljševanja medicinskih in farmakoloških terapij trenutno bolniki z Marfanovim sindromom živijo v povprečju do 60 let.
Znaki in simptomi
Če želite izvedeti več: Simptomi Marfanov sindrom
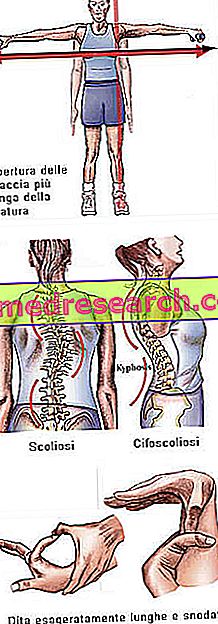
Marfanov sindrom lahko deluje popolnoma asimptomatsko. Prizadeti bolniki imajo pretirano vitko strukturo, ki je nesorazmerno visoka in tanka. Spodnji in zgornji udi imata veliko večjo dolžino kot deblo (dolikostenomegalija). Govori se tudi o arachnodactyly, ki najbolje izraža koncept pretirane dolžine prstov, značilnega za subjekte, ki jih je prizadel Marfanov sindrom: roke so torej primerjane z nogami pajka.
Glede višine imajo ti bolniki povprečno višino nad 97. percentilom.
Med posebnimi značilnostmi, ki jih pogosto najdemo pri bolnikih z Marfanovim sindromom, so:
- Odprte roke večje od višine
- Zrahljani sklepi → pretirana gibljivost sklepov
- Deformacija prsne stene
- Kristalni premik
- Zgornji del telesa je manj razvit od spodnjega dela
- Spontani pnevmotoraks (11%)
- skolioza
- Kožne strije na stegnu, hrbtu, deltoidni, prsni ravni
Med najbolj problematičnimi znaki, ki so povezani z Marfanovim sindromom, omenimo prolaps srčnega ventila in nezadostnost mitralne zaklopke: podobno stanje lahko zlahka spodbudi dilatacijo aortnega obroča in disekcijo aorte.
Tabela prikazuje znake, ki jih lahko najdemo pri bolnikih z Marfanovim sindromom. Opisani liki niso vedno prisotni, vendar jih je mogoče najti dober del.
| Prizadeta anatomska lokacija | Možni simptomi |
Cute | Strije v prsni, ledveni in sakralni površini |
oči | Okvara vida, astigmatizem, odvajanje mrežnice, glavkom z zaprtim zakotjem, izpah leče, kratkovidnost |
Struktura kosti | Artralgija, kifoskolioza, dolicostenomelija (prekomerni razvoj v dolžini udov glede na trup), hipermobilnost, visoko nebo, deformirana prsna koša, ploske noge, ozki in tanki zapestji, nenormalno ponovni vstop / izboklina na prsnici, skolioza, ukrivljena ramena, spondilolisteza |
Dita | arachnodactyly |
pljuča | Spontani pnevmotoraks, dispneja, idiopatska pljučna obstruktivna bolezen |
Spremembe obraza | Ogivalno nebo (malformacija okusa), mandibularna retrognatija (napaka v razvoju čeljusti), podaljšan obraz |
srce | Angina pektoris, aneurizma abdominalne aorte, srčna aritmija, dilatacija / raztrganje / disekcija prsne aorte, aortna insuficienca, prolaps mitralne zaklopke |
jezik | Težava z jezikom |
diagnoza
Glede na več kot 200 možnih mutacij je uporaba genetskih označevalcev skoraj nemogoča za diagnostične namene.
Ugotovitev Marfanovega sindroma ni vedno tako takojšnja, saj fenotipska ekspresija mutacije ni vedno očitna in preprosta za identifikacijo. Diagnostična zakasnitev lahko resno ogrozi preživetje bolnika: pomislite na primer na neuspeh pri prepoznavanju kardiovaskularnih težav.
Diagnostična merila za Marfanov sindrom so bila pripravljena na mednarodni ravni leta 1996: diagnoza je sestavljena iz preiskave družinske anamneze, povezane s kombinacijo večjih in manjših indikatorjev sindroma.
Nekateri od številnih uporabljenih diagnostičnih testov so:
- ehokardiogram
- Magnetna angiorezonanca in CT (za preiskavo aorte)
- magnetna resonančna angiografija (MRA) z kontrastno tekočino (za ugotavljanje notranjih struktur aorte)
- pregled s špranjskimi svetilkami (za analizo možne dislokacije leče)
- merjenje očesnega tlaka (da se poudari možna prisotnost glavkoma)
- genetski testi (priporočljivo pred spočetjem otroka, da se preveri sindrom ali ne)
terapije
Ker gre za genetsko bolezen, ni nobenega zdravila ali zdravljenja, ki bi lahko obrnilo bolezen.
Uporaba zdravil je kljub temu bistvena za ublažitev simptomov in preprečitev kakršnih koli zapletov, zlasti srčnih. V ta namen so zlasti primerna zdravila za znižanje krvnega tlaka, kot so sartani (zlasti), zaviralci ACE in zaviralci adrenergičnih receptorjev beta.
V okviru Marfanovega sindroma lahko pacienti, ki trpijo zaradi skolioze, sledijo posebni oskrbi, pa tudi tistim, ki jih je prizadel glavkom.
Operacija je možna, da popravimo nepravilno dilatacijo aorte, ki pogosto združuje večino bolnikov z Marfanovim sindromom.
Naprej: Marfanov sindrom - droge in nega »


